Pipeline overview
Jasen is a pipeline for resistance, virulence and epityping of whole genome sequenced infectious bacteria. The pipelines is intended to be used in clinical routine and therefore contains a currated, and opinionated, set of softwares and methods for assessing the samples quality.
The pipeline is divided into workflows dedicated for analysing one or a set of related species, for example can the Escherichia coli workflow also be used for analysing Shigella. See the documentation on the individual workflow for information on how it can be applied. Analysis for most of the species follows general bacterial workflow (see below[1]) with additional species-specific analysis.
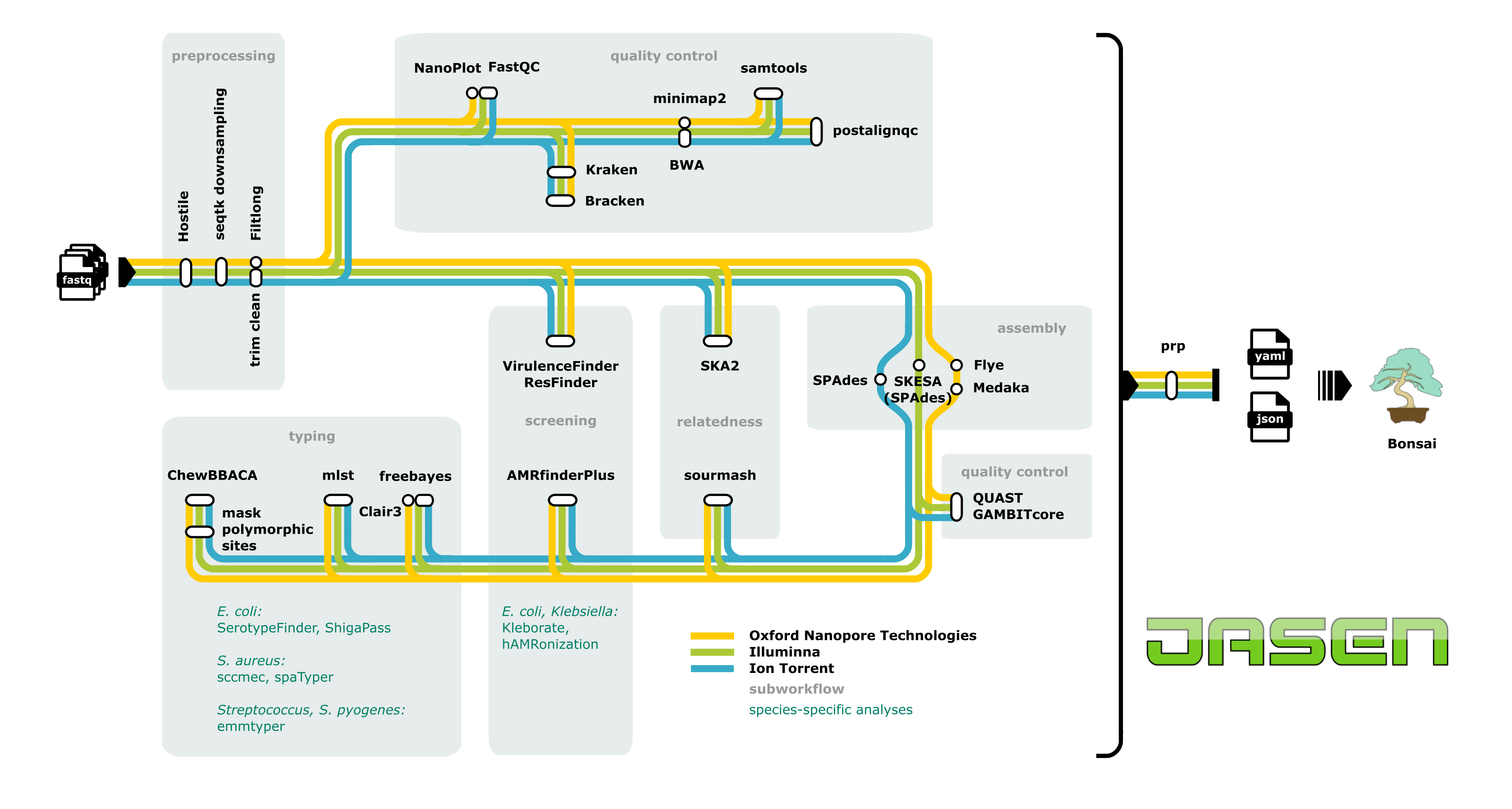
Flowchart depiciting the main workflow in Jasen, where each line represents a sequencing platform-specific workflow. Species-specific analyses are listed in respective subworkflow. Results from all the tools are collected in final json file and can be viewed in Bonsai, a visualisation tool created to explore Jasen output. Note: Mycobacterium tuberculosis analysis follows a distinct workflow and is not included in this visualisation. Masking of polymorphic sites has not been tested for ONT and is turned off by default, however it is possible to activate it in the config file.
Status on workflow development
Jasen is being continiously developed with the aim to add support for more species. Therefore can workflows be at different stages of development and evolve at different pace. A workflow can be either of three stages (development, draft, and stable) of development, these reflect the stability and the likelyhood of radical canges being made to the workflow.
Workflows under development are being actively worked at. While all workflows should be functional in the masters branch and for every release, there are no guarantee that workflows under development produce accurate results and major changes can be introduce without notice. Use a your own discretion.
Draft workflows are considered feature complete and in the process of being validated. Software versions can still change without notice. Use with discretion.
Stable workflows are feature complete and have been validated for introduction into clinical routine by one or more partners. Updates to these workflows, and versions of softwares used by these workflows, are introduced periodically after a discussion in the Jasen community.
Workflows
Species |
Development status (Illumina) |
Development status (ONT) |
|---|---|---|
Stable |
Draft |
|
Stable |
||
Stable |
||
Draft |
Draft |
|
Stable |
||
Stable |
Input
Jasen is designed for analysis of sequenced whole genomes from bacterial isolates. The pipeline takes short-read (Illumina, Ion Torrent) or long-read (ONT) sequenced reads, in compressed FastQ format, as input.
The paths to the reads are defined in a CSV file with sample id, and optionally sequencing run and lims id. See the usage page for information on how to start an analysis.
Output
Jasen will publish the analysis result to the path specified by the outdir variable in the config file. The pipeline will combine the output of the different softwares into standardised result file in JSON format for easier downstream processing of the result. The combined output have the same format regardless for workflow and can be uploaded to the result visualisation tool Bonsai for easy analysis. See the usage page for information on output.
The pipeline will also publish the output files of every tool beign run seperatly in the outdir folder.